Global 2023-01-09
Latest ISO 10993 Standards and Guidelines for Biocompatibility Evaluation of Medical Devices and Medical Packaging
LEON Biotech Co., Ltd. Kao Chih Hao
Fundamental framework of the latest revision of ISO 10993
The ISO 10993 series of standards govern biocompatibility evaluation procedures and methods for medical devices. Since the 1990s, designers and manufacturers of medical devices have been required to adhere to these critical core specifications. Since the publication of the most recent ISO 10993-1:2018 standard by the International Organization for Standardization in 2018, the various ISO 10993 series of standards have undergone significant changes which have a significant impact on the compliance with laws and regulations governing the medical device industry. To evaluate the biosafety of products through risk assessment and management, the latest version of ISO 10993-1 focuses on the entire medical device product, with the areas of focus now including materials, related manufacturing procedures, and residual substances resulting from the manufacturing process.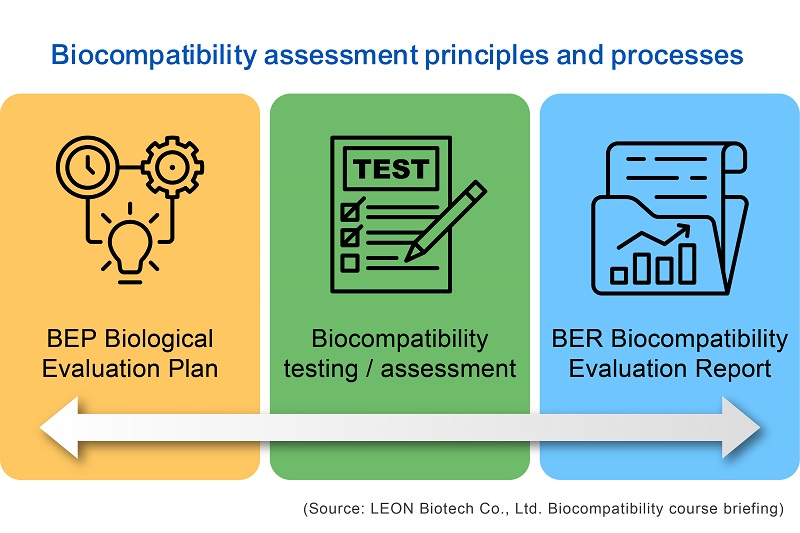
The latest version of ISO 10993-1 requires manufacturers of medical devices to prioritize material traceability and data collection integrity, as well as apply risk assessment procedures from ISO 14971 to the selection and use of all materials. According to the most recent ISO 10993 requirements, prior to implementing the overall biocompatibility assessment report, biocompatibility assessment reports for various products must be conducted based on the PDCA (Plan-Do-Check-Act) concept in the quality system procedure (Figure 1).
Manufacturers or developers of medical devices must propose corresponding assessment plans and expected implementation process in the early stages of design and development, as well as establish the product's safety acceptance criteria before conducting corresponding testing or assessment operations according to their plans. The goal is to generate comprehensive assessment reports in accordance with risk assessment principles based on collected information or tests. The risk assessment principle from ISO 14971 is much emphasized in the latest version of ISO 10993-1 to carry out a series of biosafety factor assessments which include biological hazard identification, determination of harmfulness to the human body, or the use of qualitative and quantitative analysis and test results of composition and leachability to validate the safety of the product.
The Priority and Emphasis of chemical characterization in ISO 10993-18
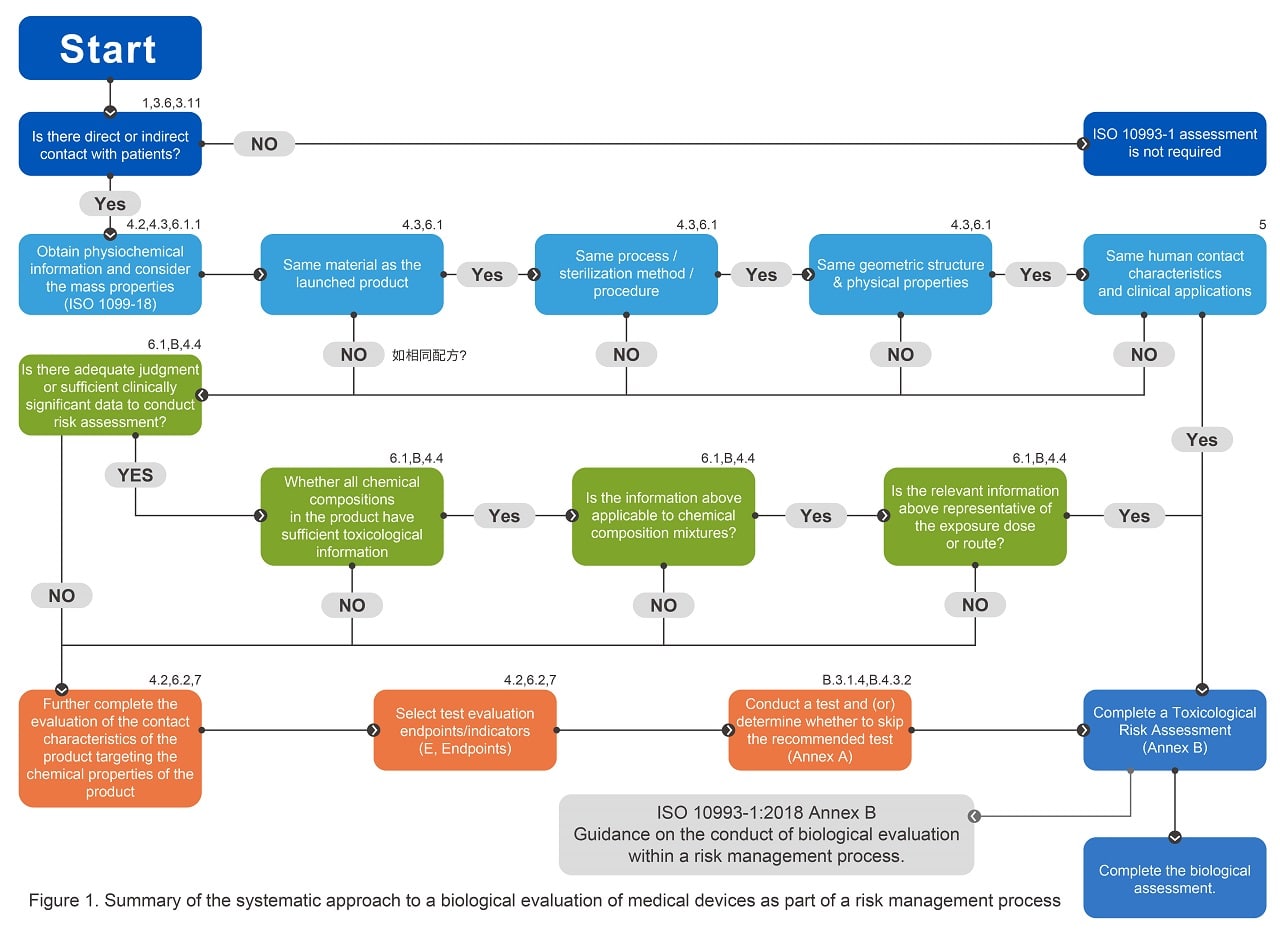
The latest version of ISO 10993 clearly indicates in the assessment process (Figure 2) that the first step in assessment is to conduct chemical characteristic analysis and data collection. This is also the most significant change in the review of standards adopted by competent authorities since the publication of the latest version of the ISO 10993 standard.According to the standard description for ISO 10993-18, the requirements are primarily to collect relevant information on the product's "material composition," "residues from the manufacturing process", as well as "extractables/leachables” - in particular the extractables/leached substances - to determine whether they are hazardous or not. In accordance with the recommendations in ISO 10993-18, inorganic leachable, non-volatile organic compounds, and volatile organic compounds will be qualitatively or quantitatively analyzed. Currently, sophisticated chemical analysis mass spectrometry equipment such as inductively coupled plasma mass spectrometry (ICP-MS), liquid chromatography mass spectrometer (LC-MS), gas chromatography mass spectrometer (GC-MS), and even higher-order analytical equipment, are frequently utilized to identify these compounds as the objectives are: identify any potentially hazardous substances in the product, establish an allowable limit for each detected compound using the ISO 10993-17 toxicological assessment procedure, and compare the analyzed data to safety limit requirements.
To determine the margin of safety, it is necessary to conduct a series of comparisons with existing products on the market which include an analysis of the materials' efficacy, processing geometry, and physical properties which are relevant to the product’s intended clinical application(s).
Selection and precautions for ISO 10993 biological tests
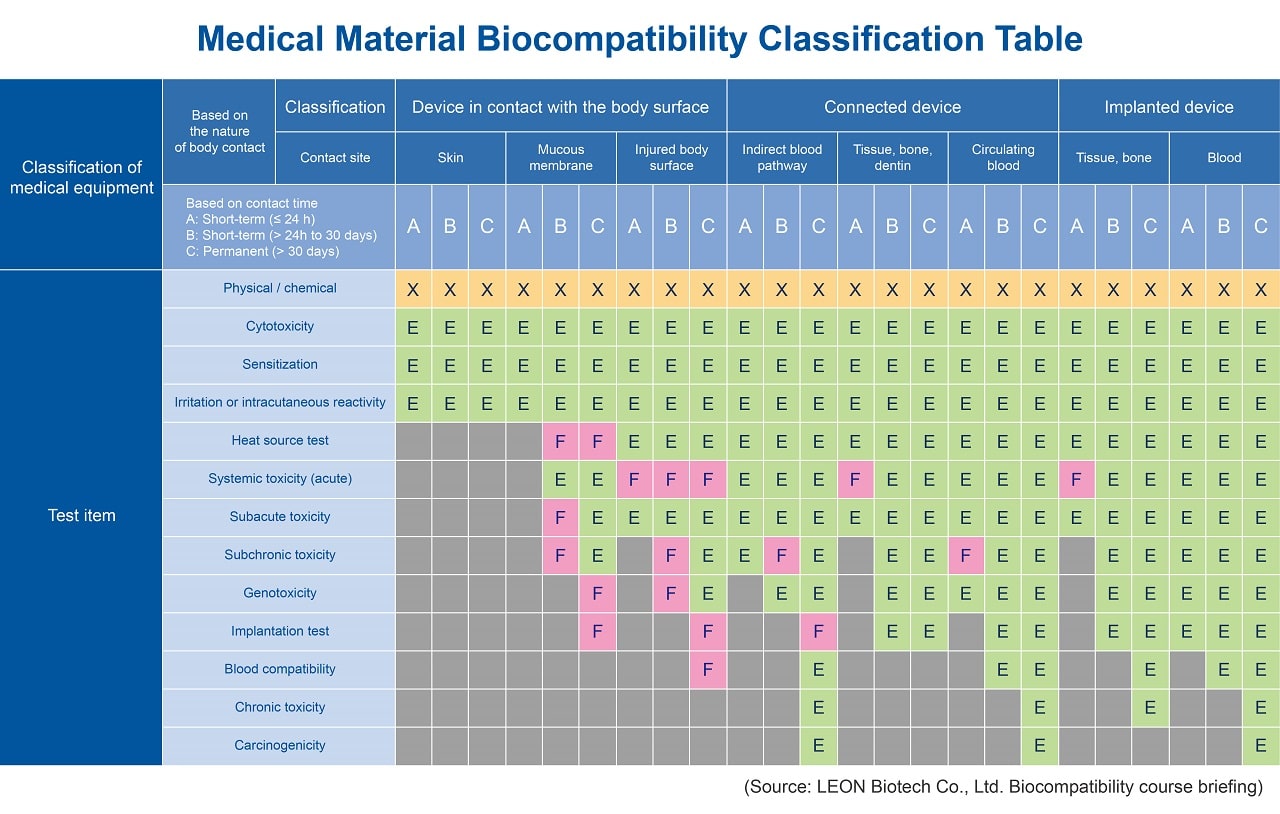
In previous version of ISO 10993, the biological tests in Table A-1 of ISO 10993-1 were used as the basis for test planning. In the latest version of ISO 10993-1, the biological tests (Table 1) are converted to become the endpoint of risk assessments, going against both the spirit of mandatory biological testing and calls for a wide range of biological indicators as the evaluation's final metric. Manufacturers of medical devices can verify compliance with the various safety requirements through the use of substantial equivalence comparisons to currently available toxicological safety data or published literature. In the absence of sufficient data, the biological tests design is used to meet the different requirements of different products. Table A-1 emphasizes the following areas:(1) The physical and chemical characteristic of the product's materials must first be collected and assessed for risks.
(2) The latest version of standard test items includes material-mediated pyrogenicity, chronic toxicity, carcinogenicity, reproductive toxicity, and degradability (new materials must be evaluated).
(3) "X" denotes mandatory biological risk assessment information, and "E" denotes the risk assessment endpoint requiring evaluation.
(4) "F" standard for US FDA specific requirements.
The selectivity and representation of the sample are primary considerations in various tests. There are a number of preparation conditions and requirements for test samples governed by ISO 10993-12. The test samples may be in the form of "final finished products (sterile)," "representative samples with multiple specifications, or simulated representative samples produced using the same processes". The test sample is prepared according to the specified extraction conditions, which are in accordance with ISO 10993-12. Test samples are prepared and include various extraction conditions (temperature and data) and extracted solution ratios. This procedure should also be evaluated and selected based on the characteristics of the product material.