全球 2022-12-26
醫療器材與包裝材 ISO 10993生物相容性最新要求趨勢
撰文:亮宇生物科技股份有限公司 高志豪
新版ISO 10993基本架構
ISO 10993系列標準規範了醫療器材產品的生物安全性評估程序與方法,自90年代開始一直都是醫療器材設計製造業者依循的重要安全性核心規範。自從2018年ISO組織頒布最新一版的ISO 10993-1:2018起,至今陸續在相關10993各系列標準都有重大變更,對於醫療器材行業的法規依循有了莫大的影響。新版的ISO 10993-1針對整體的醫療器材產品包含在材料本身、相關的製造程序以及製程的殘留物質以風險評量與管理的方式評價產品的生物安全性。要求醫療器材業者更加著重在材料的追溯性與資料收集的完整性。並貫徹另一主幹標準ISO 14971的風險評價流程套用於整體材料的選擇與使用。目前依據ISO 10993的最新要求,執行整體生物相容性風險評估報告前,須依據品質系統程序PDCA(Plan-Do-Check-Act) 的概念進行各產品的生物相容性評估報告。(如右圖) ISO生物學測試之選擇與注意事項
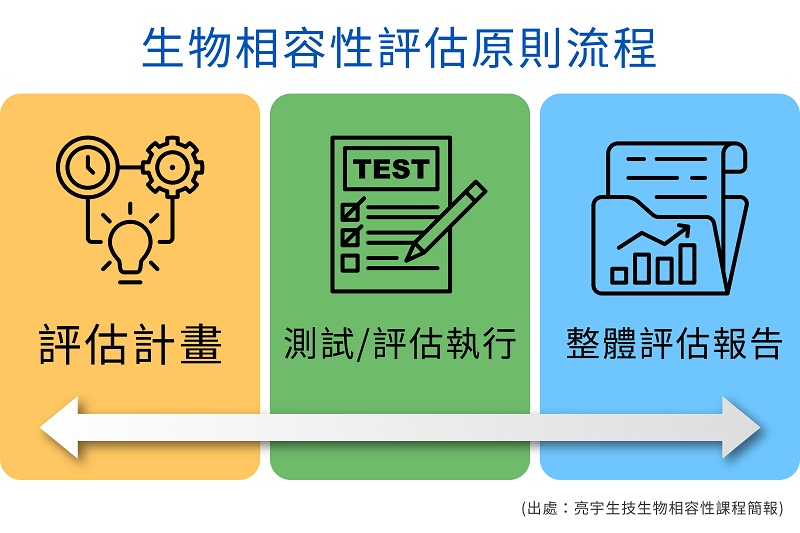
醫療器材製造廠或是開發者必須在設計開發前期擬定相對應的評估計畫與預計的實施流程,設定該產品對應的安全性允收規格,並且依據計畫內的規劃進行對應的測試或是評估作業,最終將所收集的資訊或是測試整合依據風險評估的原則進行完整的評估報告。在新版的ISO 10993-1強調採用ISO 14971的風險評量概念,進行一系列的生物安全因子的評估,包含生物性危害的鑑別,評斷其對於人體的危害性,並且採取適當的定性與定量分析與測試結果來證明其組成含量或是溶出量的安全性。
ISO 10993-18化學表徵之優先性與重點
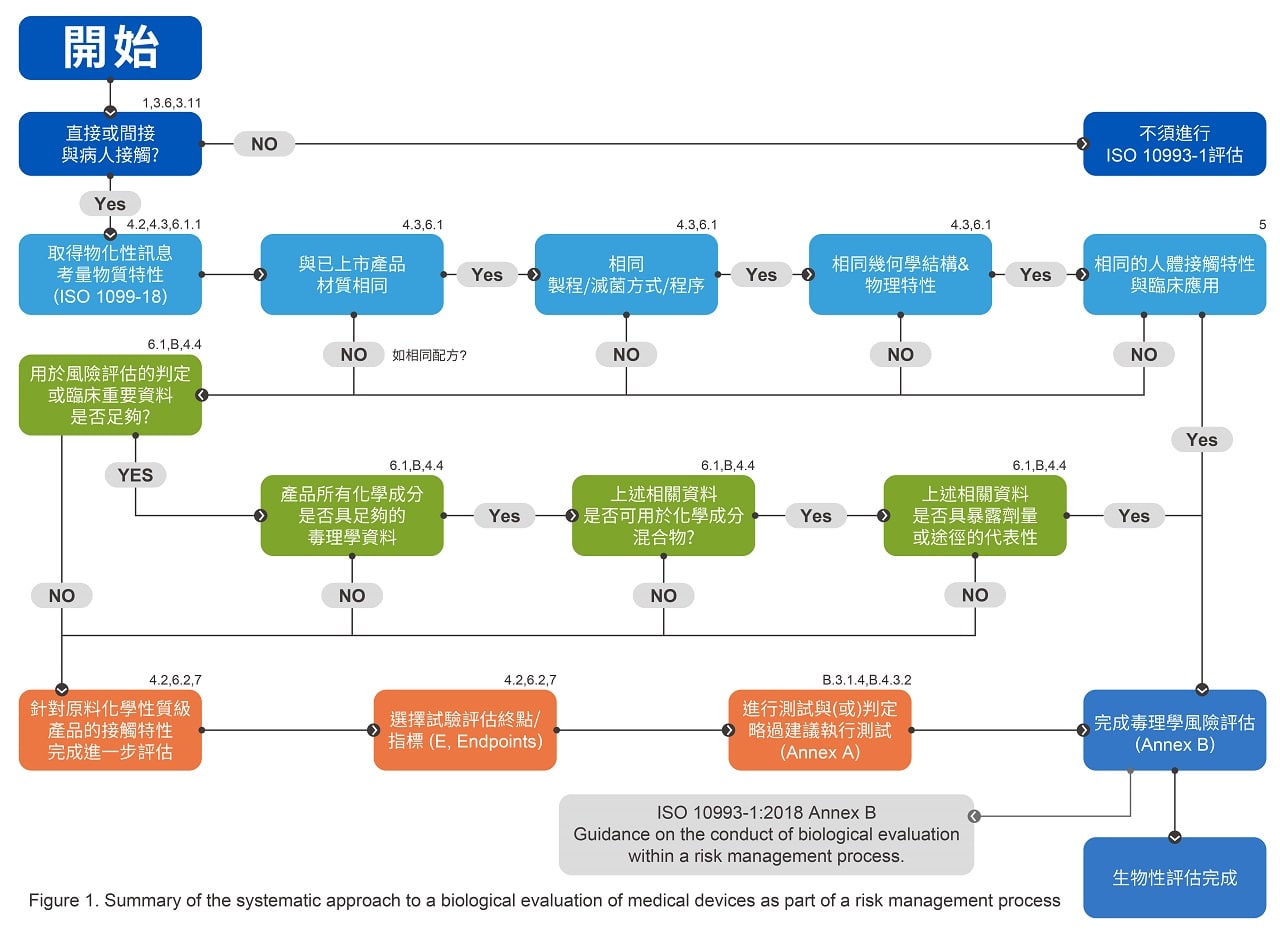
新版ISO 10993在評估流程(如下圖)中開宗明義要求評估起始點要進行物理化學特性的分析與資訊收集,這也是新版標準頒布以來各國主管機關採用標準的最大審查變化,此要求根據ISO 10993-18的標準描述,主要是要針對產品的「材料組成物」、「製造程序中的殘留物」還有「可能的萃取物(Extractable)/溶出物(Leachable)」收集相關的資訊,其中又特別針對所謂的萃取/溶出物質需要採用毒理評估方式對其鑑別風險危害與否。依據ISO 10993-18的規範建議,會針對無機溶出物、非揮發有機物與揮發性有機物進行定性或定量分析,目前坊間多採用高階的化學分析質譜設備來進行鑑別,例如感應耦合電漿質譜儀(ICP-MS)、液相層析質譜儀(LC-MS)、氣相層析質譜儀(GC-MS)甚至更高階的分析設備。目的都是為了找出產品中可能的危害風險物質,並以ISO 10993-17的毒理評估流程設定每一個檢出化合物可接受的允許限量,將分析的數據比對限量允收的要求進行安全限值的計算(margin of safety),並且需要進行一連串的比對已上市產品的比較,包含材質、加工製程、幾何構形及物理性質與臨床預期用途對應的有效性分析。
創造雙贏,大於產品價值的服務價值
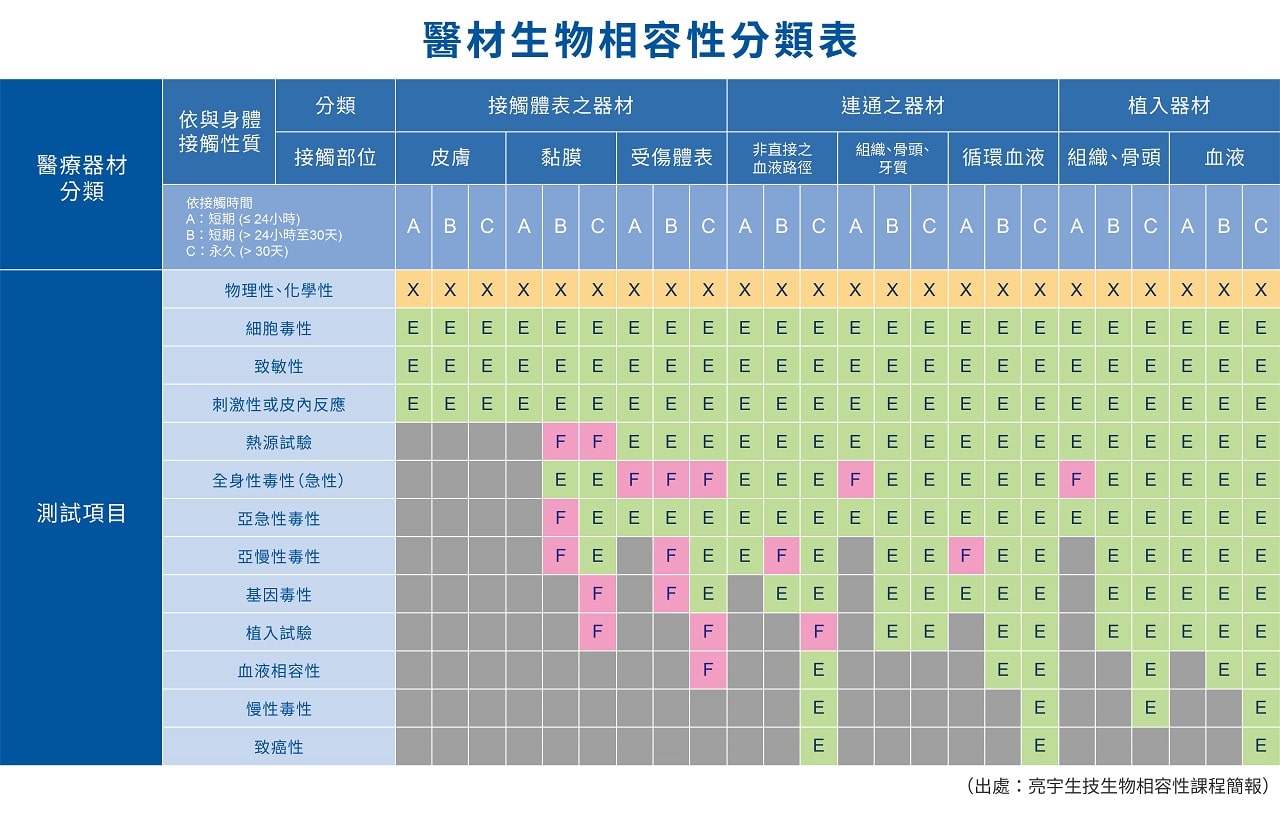
在舊版的ISO 10993中,常以ISO 10993中表A-1的各項生物學測試當作測試的規劃依據,而在新版的要求中,各項生物學的測試(表一)轉變成風險評估的終點指標(end point),不再以強制要求生物測試為目的,改以要求各項生物學指標做為評估終點,醫療器材業者可以透過實質比對,既有毒理安全資料或是文獻來佐證安全性的各項要求,若無可依循的資料下,才以最終設計生物學測試的手段來滿足不同產品的不同需求。表格著重以下重點:
(1) 產品選用材料的物理性及化學性資料必須先收集及進行風險評估
(2) 新版標準的測試項目增加了熱原性(Material Mediated Pyrogenicity)、慢毒(Chronic toxicity)、致癌性(Carcinogenicity) 、生殖毒性及可降解性(新材料需評估)
(3) “X”表示生物學風險評估的前置信息,”E”表示要在風險評估中評估的終點
(4) “F”則為美國FDA 的特定要求
各項試驗中需要優先注意的是樣品的選擇與代表性,ISO 10993-12針對測試樣品有規範幾種樣品製備的條件與要求,測試樣品來源可以來自於「最終成品(無菌)」、「多種規格下代表性樣品」以及「經由同樣製程產出之模擬代表樣品」。並且經由指定的萃取條件進行試驗物質的準備。包括依據不同的萃取條件(溫度與時間),還有與萃取溶液的比例)進行測試樣品的製備。這部分也要依據產品材質的特性做評估與選擇。
醫療包材生物相容性之要求
在新版ISO 10993-1的標準也有提到須將與產品接觸的包裝材料列入評估的一環,主要是因為產品會長時間與包裝材料接觸,需考量包裝材料是否會產生溶出物質或是遷移性的物質移轉到醫療器材上,因此對於包裝材料的化學分析有進行評估的必要性。另外也須針對會直接接觸醫療器材的包裝材料要進行生物安全性的評估,相關資訊可參考ASTM F2475-20之準則;此外,若是醫療器材用於藥品包裝上則需要依循USP<87>Biological Reactivity tests, in vitro , USP<88> Biological reactivity tests, in vivo, 進行Class I~IV因應侵入性或是植入性產品的材料安全性測試,用此以降低整體的產品風險。另外包裝材料的潔淨度也跟產品的潔淨度息息相關,所以在微粒殘留上也須要多考量。
總結
醫療器械的風險無所不在,伴隨著本次10993-1的改版對於醫療器械風險管理的要求提高,各級醫療器材生產商更需要重新在產品生命週期(Product life span)檢視生產各階段過程中可能殘餘的風險物質。而對於產品委託具有GLP協力廠商資質的實驗室和測試過程中的檔案保存也更趨重要,如果沒有在上市前收集產品材料資訊確認其安全性和降低風險,到臨床醫院端引起病患不良反應的後果將難以想像,這對於生產廠家的品牌聲譽及信心度更會造成劇烈的影響。藉由本次10993-1改版將可使醫療器械的風險評估架構更趨完整,也符合近年來各國對於醫療器械監管的力道趨於嚴謹,在近期新版的歐盟MDR法規與美國US FDA對於生物相容性的要求我們也能同步看到。隨著後疫情時代健康產業的蓬勃發展,更多新型態新用途高風險的器械設計或生醫新材料的開發將透過合乎法規的態度落實在臨床使用的安全性上。